paired end sequencing r1 and r2
So while R1 starts with your 5 adapter for R2 anything is possible depending on the length of the fragment of interest and the read length. Adapter trimming can remove FASTQ sequences if the.
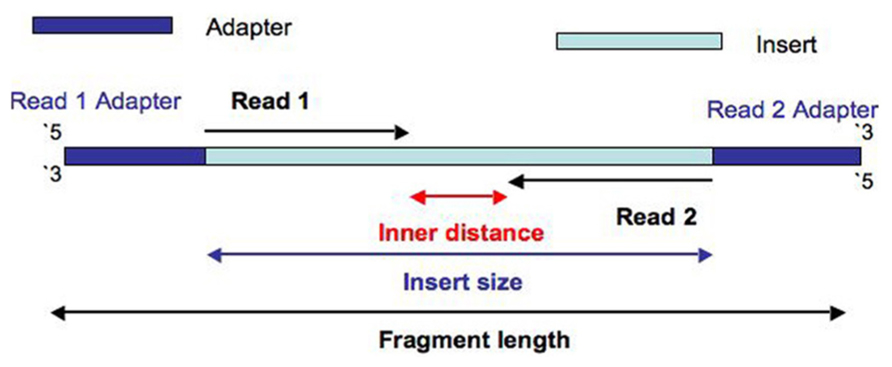
How To Quantify The Overlapping Reads In Paired End Dna Sequencing To Check The Sequencing Efficiency
Modified 4 years 8 months ago.
. The Illumina paired-end sequencing technology can generate reads from both ends of target DNA fragments which can subsequently be merged to increase the. If the sequencing sample is the same as the original copy the read R1 should be mappable in. My understanding is that paired end reads from the Illumina HiSeqMiSeq platforms looks something like this.
The steps below can be used if. If youre running MiSeq Reporter 23 theres a new setting called StitchReads that can do this for you. The map is 80 but the R1 R2 counts are.
The 2 reads should overlap in middle. Because GBS-SNP-CROP utilizes paired-end reads the total number of actual reads used R1 and R2 is twice this number d The total number of nucleotides of sequence data. This is quite common in single-cell RNA-seq.
The amplicon was 550nt sequenced using 300nt x2 Illumina chemistry. For each cluster that passes filter a single sequence is written to the corresponding samples R1 FASTQ file and for a paired-end run a single sequence is also. There are two FastQ files generated in an Illumina paired-end reads sequencing run.
Where xxx is a. Simply add StitchReads 1 to your Settings. The files have this naming convention.
In conventional paired-end sequencing you simply sequence using the adapter for one end and then once youre done you start over sequencing using the adapter for the other. As indicated in the comments yes you can definitely tell standard Illumina sequencers to sequence mates in a pair to different lengths. First sequencing can either be single-end where each sample has only sequence data or paired-end where each sample has two sequence data R1 and R2.
So it is an alignmerge task. You could see the reverse. Are paired end reads reverse.
This usually does not happen for PE sequencing which makes me a bit nervous to proceed with analysis. Download scientific diagram The direction and positional order of the paired-end reads R1R2. For paired-end alignment aligners want the R1 and R2 fastq files to be in the same name order and be the same length.
If the sequencing sample is the same as the original copy the read R1 should be mappable in forward direction in 5-end and R2 in reverse direction in 3-end. I need to merge R1 and R2 Illumina paired end reads. 10-09-2013 1013 AM.
Ngmerge Merging Paired End Reads Via Novel Empirically Derived Models Of Sequencing Errors Bmc Bioinformatics Full Text
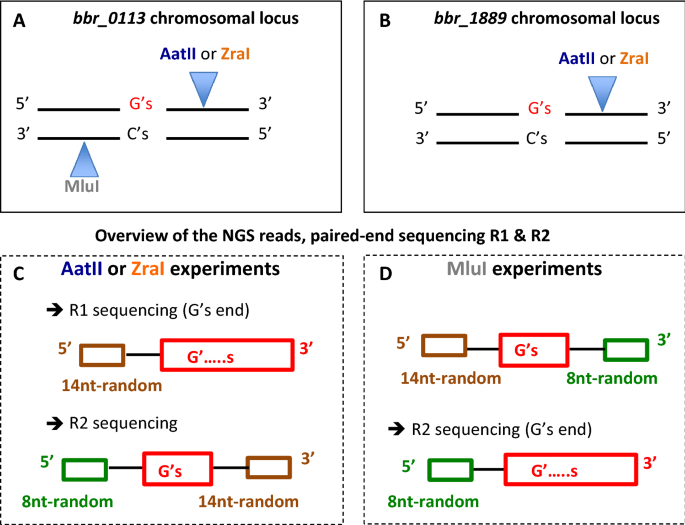
Maximum Depth Sequencing Reveals An On Off Replication Slippage Switch And Apparent In Vivo Selection For Bifidobacterial Pilus Expression Scientific Reports

Trouble With Merging Paired End Reads User Support Qiime 2 Forum
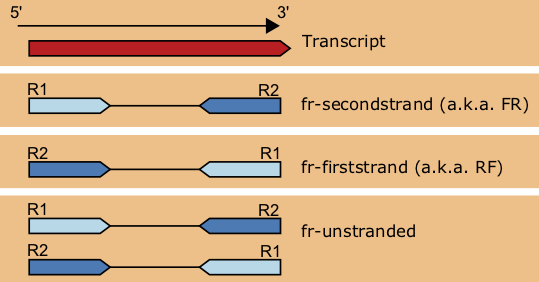
Chapter 6 Transcriptomics Applied Bioinformatics
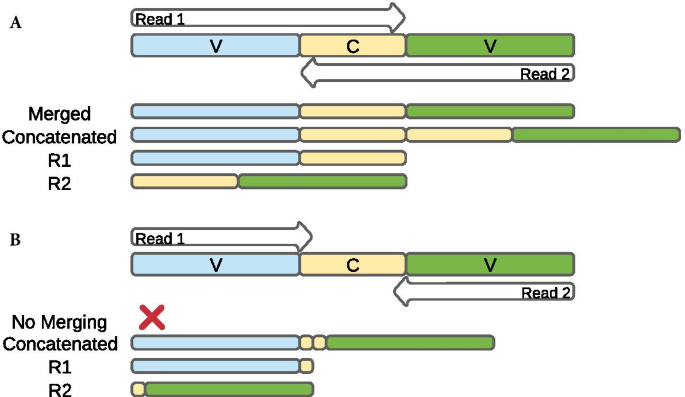
Concatenation Of Paired End Reads Improves Taxonomic Classification Of Amplicons For Profiling Microbial Communities Bmc Bioinformatics Full Text
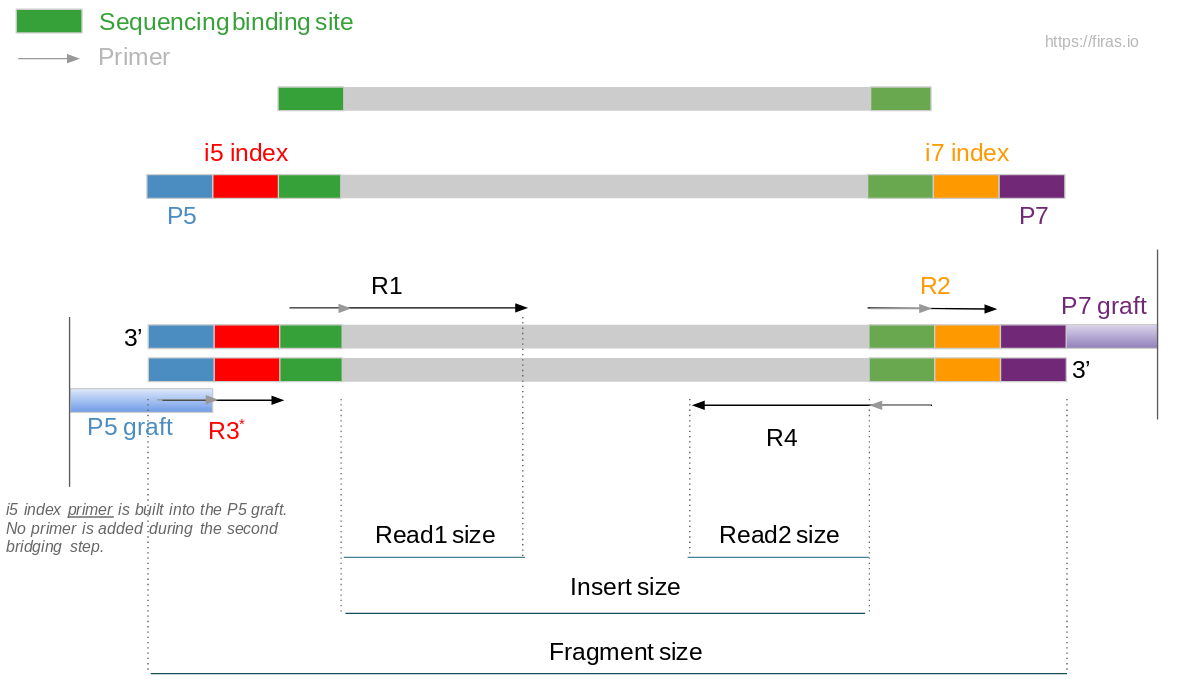
An Overview Of Illumina Multiplexing Firas Sadiyah
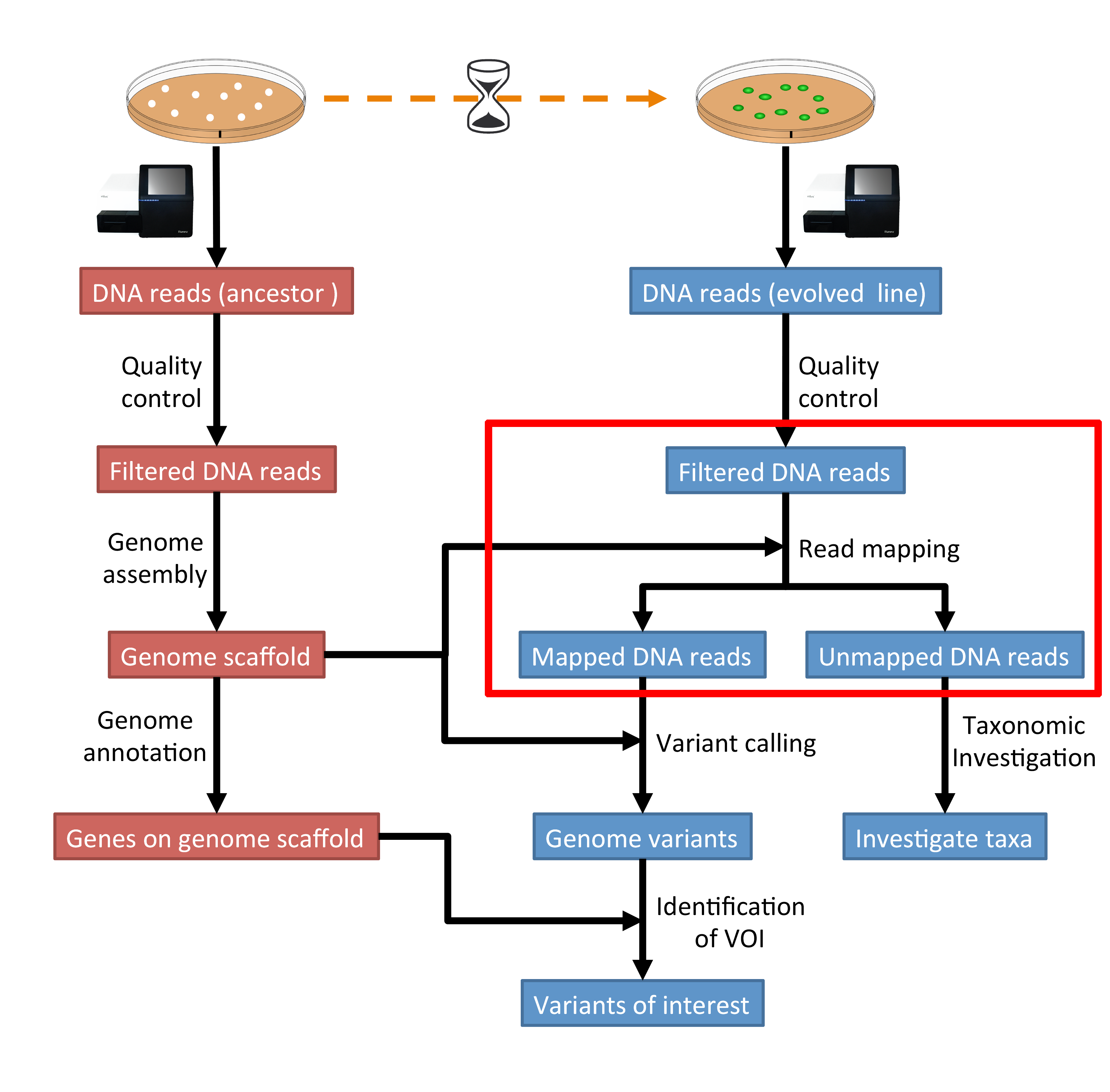
5 Read Mapping Genomics Tutorial 2020 2 0 Documentation
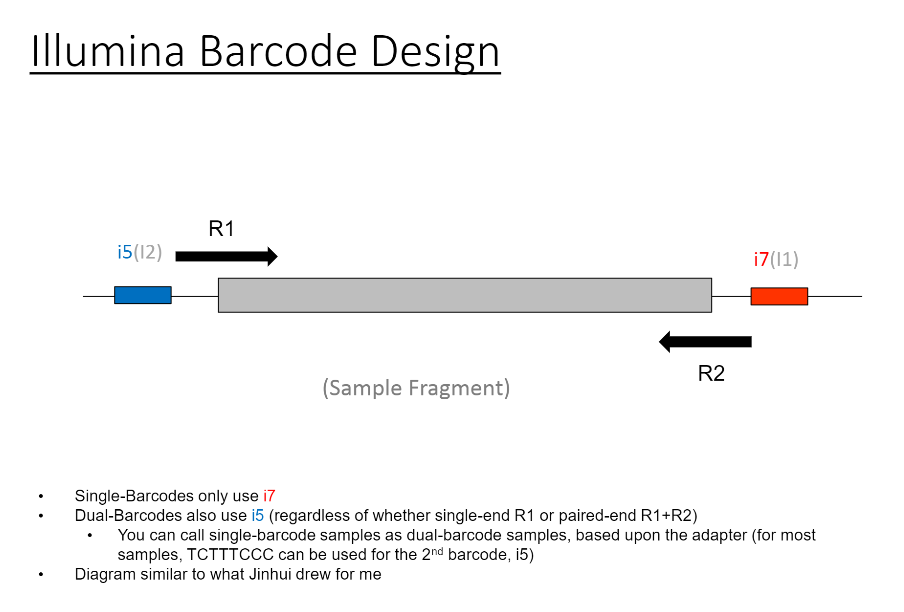
Calling Single Barcode Samples From Mixed Runs As Dual Barcode Samples Possible Illumina Run Qc Flags
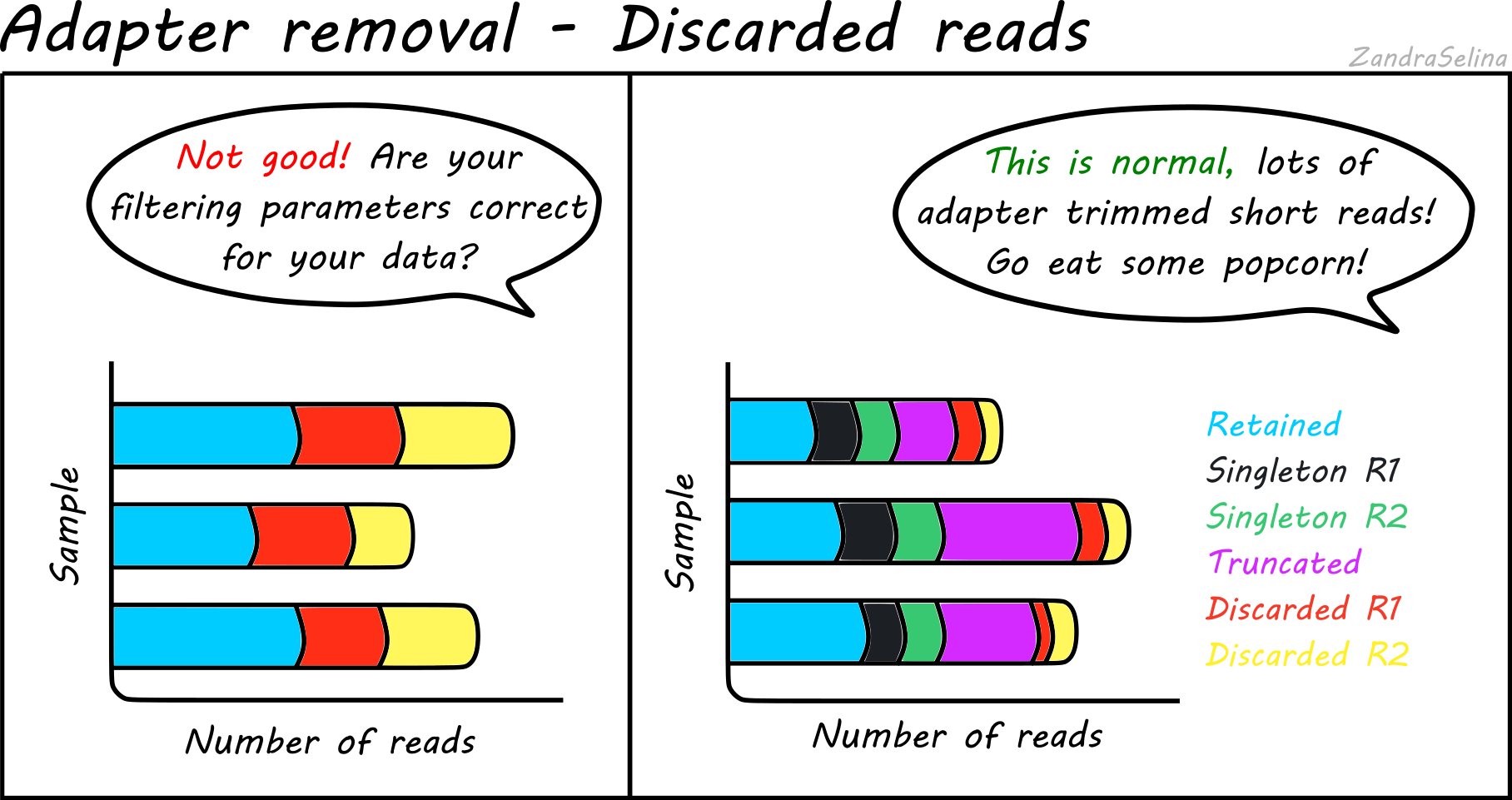
Eager Nf Core

5 Processing Raw Scrna Seq Data Analysis Of Single Cell Rna Seq Data

Epitranscriptomic Mapping Of Rna Modifications At Single Nucleotide Resolution Using Rhodamine Sequencing Rho Seq Star Protocols
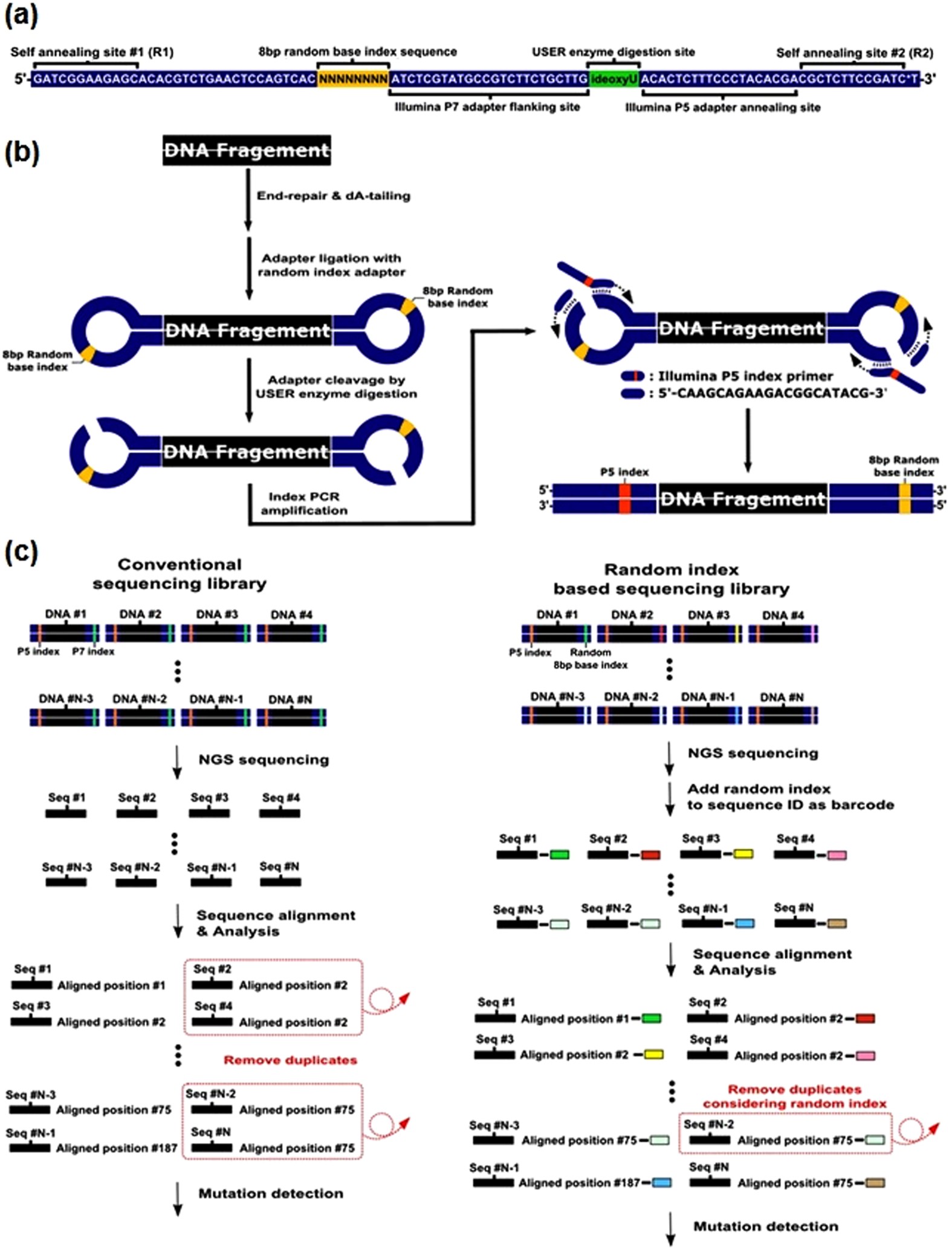
Asymmetrical Barcode Adapter Assisted Recovery Of Duplicate Reads And Error Correction Strategy To Detect Rare Mutations In Circulating Tumor Dna Scientific Reports

How I Will Import Combined R1 Fastq And R2 Fastq Files Into Qiime2 User Support Qiime 2 Forum
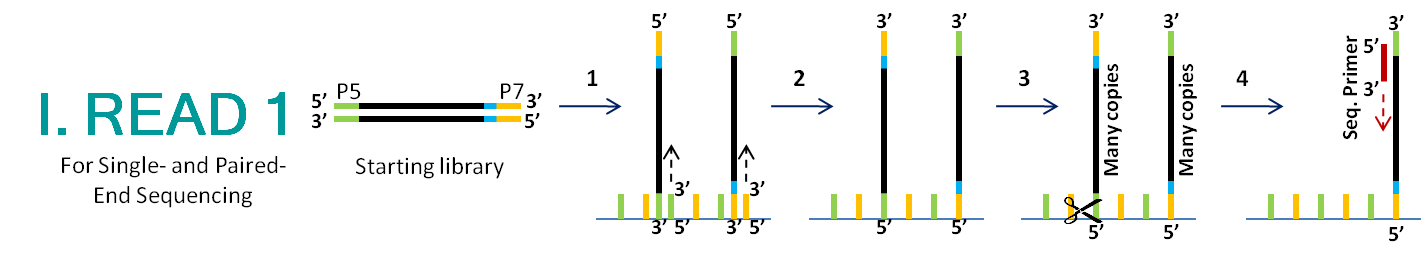
The Mgh Nextgen Sequencing Core Core Services

Dmx With 4 Initial Read Files I1 I2 R1 R2 User Support Qiime 2 Forum
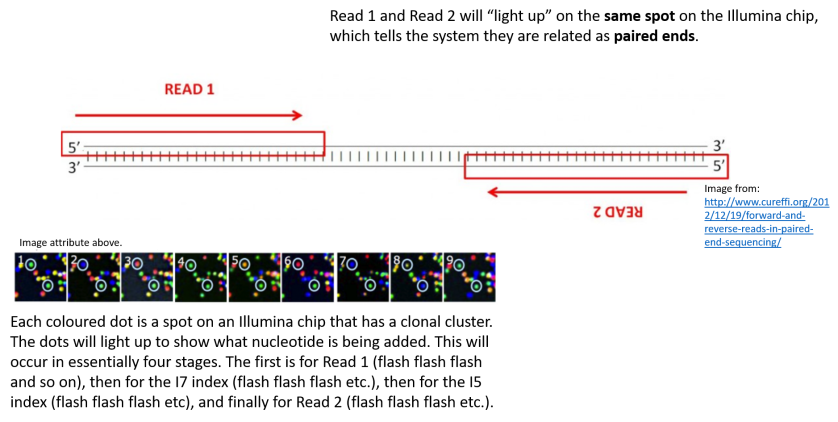
Illumina Sequencing For Dummies An Overview On How Our Samples Are Sequenced Kscbioinformatics
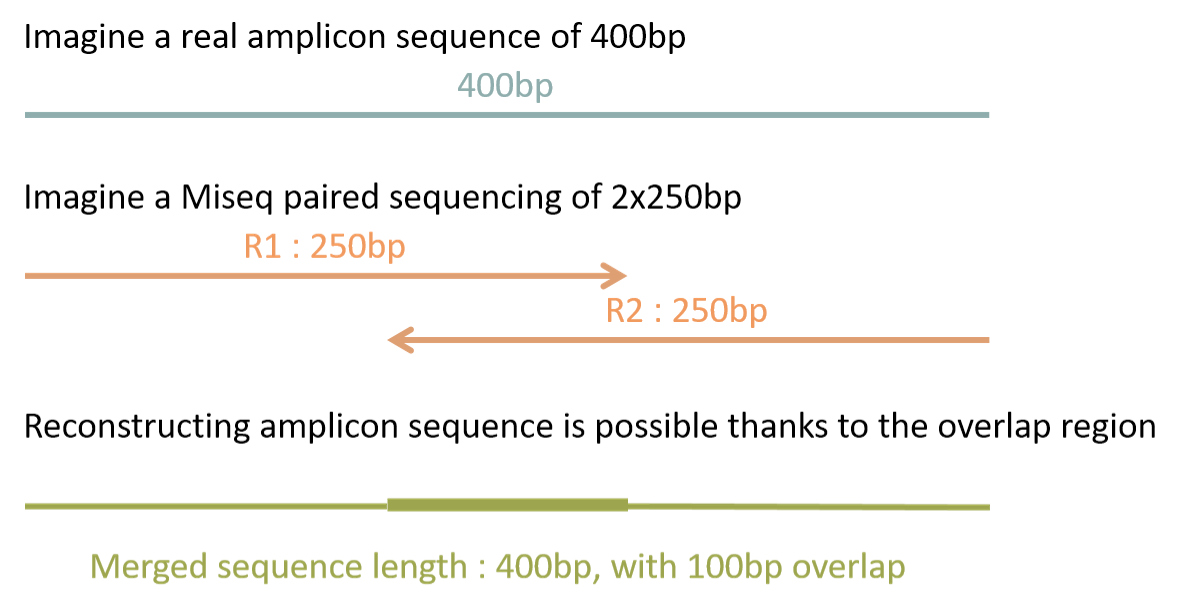
Frogs Faqs
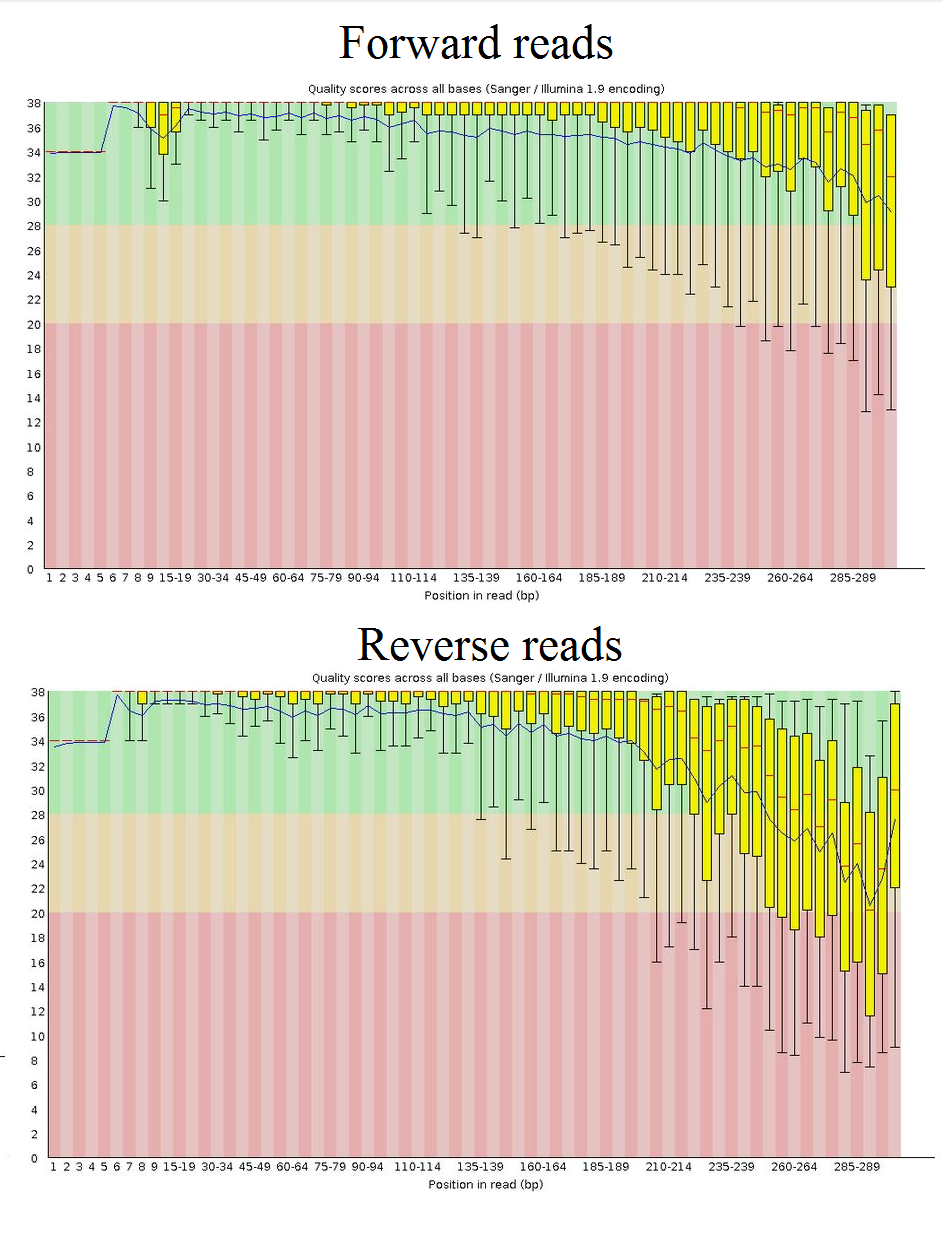
16s Rrna Analysis
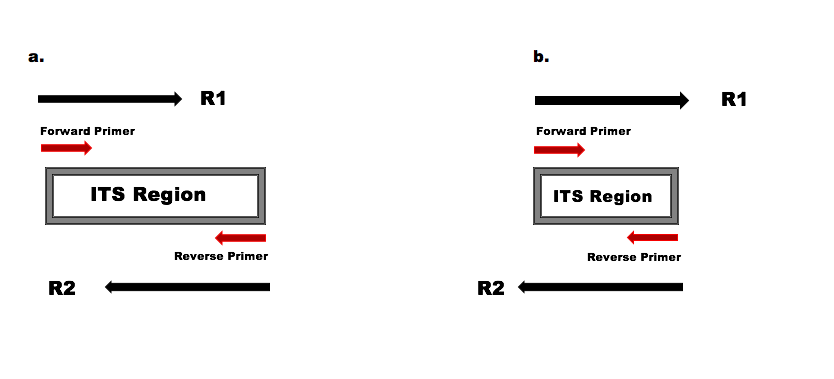
Dada2 Its Pipeline Workflow 1 8